| Měnící se tvář parkinsonské neurodegenerace |
|
prof. MUDr. Petr Kaňovský CSc. |
| SANQUIS č.84/2010, str. 96 |
Parkinsonova nemoc bude brzy 200 let stará. Poprvé totiž vědci toto neurodegenerativní onemocnění popsali v roce 1817, nikoli však pod jejím nynějším jménem, ale pod názvem „shaking palsy“, který by se dal do češtiny přeložit asi jako „obrna třaslavá“. Takto mimochodem byla nemoc uváděna v neurologických učebnicích ještě v době Hennerově.
|
|
Název Parkinsonova dostala nemoc od Jeana-Martina Charcota až o 60 let později. Mimochodem, James Parkinson nebyl neurolog (tato specializace v té době pochopitelně neexistovala), ale praktický lékař, ordinující na nepříliš noblesní adrese Hoxton Square v severovýchodním Londýně. Dnes se tu nachází „trendy“ galerie moderního umění White Cube, jež patří Charlesovi Saatchimu; a i jinak jde o adresu zajímavou, neboť ve zdejších studiích začínal kariéru Alfred Hitchcock a právě tady svou první restauraci otevřel Jamie Oliver.
James Parkinson neordinoval denně, neboť ordinaci zdědil po otci, klinická práce jej příliš nebavila a spoustu času věnoval politické práci – sepisování pamfl etů proti králům Jiřímu III. a Jiřímu IV. a premiérovi Williamu Pittovi mladšímu. Za to byl koneckonců spravedlivě několikrát vyhoštěn do Skotska, kde dobu vyhnanství vždy trávil v nehostinném Edinburghu. Patrně tedy nepřekvapí, že ani šest nemocných, jejichž pozorováním vznikl popis „třaslavé obrny“, nepatřilo k Parkinsonovým pacientům, ale byli to lidé, které pravidelně či nepravideně vídával oknem své ordinace „šourat se“ po Hoxton Square.
Parkinsonův popis nemoci vystihuje vlastně první, klasickou a po více než sto padesát let neměnnou tvář parkinsonské neurodegenerace. Zahrnuje dodnes uznávanou triádu příznaků: klidový třes, ztuhlost (rigiditu) a povšechné zpomalení pohybu (bradykinezi), která byla v posledních letech doplněna na tetrádu příznakem posturální instability. Parkinsonova nemoc se považovala za prototyp parkinsonského, postaru „hypokineticko-rigidního“ syndromu, a za její hlavní příčinu se označoval tzv. „status lacunaris cerebri“, kdy je oblast bazálních ganglií postižena drobnými mozkovými infarkty a mozková tkáň makroskopicky připomíná sýr s malými oky. Nutno zde poznamenat, že u poměrně (bohužel) velké části neurologické komunity zůstala tato představa parkinsonismu a jeho příčin platná dodnes.
PSP a multisystémová atrofie
Již začátkem 60. let minulého století si několik bystrých kanadských neurologů začalo všímat určitých specifik klinického obrazu u pacientů, kteří se léčili s diagnózou Parkinsonova nemoc. Patrně prvním z nich byl Jamie Olszewski, který u svého pacienta pozoroval „zvláštní“ typ pohledové obrny a absenci třesu. Byvše praktickým neurologem, který neměl další možnosti paraklinického testování, odeslal pacienta Johnu Richardsonovi, který ke konzultaci přizval dalšího kolegu, Johna Steela. Tak došlo k tomu, že první z tzv. „parkinson-plus“ syndromů se dnes kromě nicku PSP („progressive supranuclear palsy“ – PSP) nazývá nemocí Steele-Richardson-Olszewski. Je však otázkou, které z podob nemoci toto jméno náleží, neboť další vývoj poznání přinesl rozdělení nozologické jednotky na tzv. „PSP Richardsonova typu“ a „PSP parkinsonského typu“. Musíme však uvést, že John Steele na posledním světovém neurologickém kongresu sám navrhl, aby se porucha nadále klasifikovala jako jedna nozologická jednotka a aby se jmenovala Olszewskiho nemoc.
Další v řadě bylo onemocnění, které kombinovalo příznaky parkinsonismu s autonomní dysfunkcí a mozečkovým a pyramidovým postižením. Porucha s dominantní autonomní symptomatologií se již od poloviny 50. let nazývala Shy-Dragerovým syndromem, aniž kdy kdo podrobněji zkoumal charakteristiky jeho parkinsonských symptomů. Až v roce 1989 Nial Quinn pojmenoval poruchu jako multisystémovou atrofii a postuloval klinická kritéria onemocnění. Pokrok však šel i potom pomalu: v České republice zpochybňovaly autority existenci této nemoci ještě v roce 1996.
|
|
|
Demence s Lewyho tělísky
Zhruba před dvaceti lety se také začalo více diskutovat o incidenci kognitivní poruchy nebo demence u Parkinsonovy nemoci. Díky moderním dopaminergním preparátům a prakticky uniformní léčbě pomocí L-DOPA téměř u všech nemocných přežívali pacienti významně déle a bylo tedy jasné, že u řady z nich se s progresí nemoci vyvíjí i kognitivní deficit. Při důkladnějším zkoumání větších kohort se zjistilo, že až 50 % pacientů trpělo různým stupněm demence. Toto procento se v průběhu následující dekády postupně zvyšovalo až na 90 %. Zároveň bylo jasné, že část těchto pacientů manifestuje distinktní fenotyp, který sdílí mnohé znaky s Alzheimerovou nemocí. Podobně specialisté, věnující se Alzheimerově nemoci, popisovali u některých pacientů extrapyramidové příznaky, upomínající nápadně na Parkinsonovu nemoc. Tato koincidence obou fenotypů rozproudila diskuzi o tzv. alzheimerské variantě Parkinsonovy nemoci a vice versa o parkinsonské variantě Alzheimerovy nemoci. Souběžné patologické studie však ukázaly, že tento parkinsonský fenotyp odpovídá specifické patologii, při níž se tzv. Lewyho tělíska nacházejí prakticky v celém mozku.
V roce 1996 Ian McKeith zveřejnil první (dodnes platná) diagnostická kritéria této nemoci, které se dnes říká demence s Lewyho tělísky. Od klasické Parkinsonovy nemoci se liší tím, že pacienti trpí zrakovými halucinacemi a poměrně brzy se u nich vyvine progredující kognitivní porucha, která do dvou let vede k invalidizující demenci. Bohužel se demence s Lewyho tělísky příliš neodlišuje od Parkinsonovy nemoci s demencí, což je dodnes zdrojem diagnostické i klasifikační konfuze. Existuje nepříliš zřetelná dělicí linie, která odlišuje demenci s Lewyho tělísky od Parkinsonovy nemoci na základě rychlosti, s jakou se vyvine kognitivní porucha. To je v době ultrastrukturálních a genetických podkladů klasifikace chorob poněkud málo, ale nic víc k dispozici nemáme.
Kortikobazální syndromy
Zhruba ve stejné době, kdy Nial Quinn poprvé jasně definoval multisystémovou atrofii, si specialisté začali mezi pacienty trpícími parkinsonismem všímat těch, u kterých bylo nápadné jednostranné postižení těla. Klasická Parkinsonova nemoc totiž sice ve většině případů začíná jednostranně, ale během dvou až tří let dojde u naprosté většiny pacientů k stranovému vyrovnání. U některých pacientů ale stranová predilekce, a to významná, zůstává patrná po celou dobu průběhu nemoci.
Bližší zkoumání a opakované vyšetření odhalilo u těchto pacientů další poruchu, jejíž příčina tkví v postižení téže hemisféry, která odpovídá straně parkinsonského postižení. Tato porucha byla lokalizována do oblasti mozkové kůry a zrodil se termín kortikobazální degenerace. Přídatnou poruchu může představovat prakticky cokoli: myoklonus, poruchy symbolických funkcí, jako je řeč, porozumění řeči, psaní, čtení, pravolevá a prostorová orientace nebo tzv. „alien hand“ – syndrom cizí ruky, kdy si končetina „prakticky dělá, co chce“, nezávisle na vůli pacienta.
Kortikobazální degenerace slouží jako ideální příklad toho, jak dále dochází ke štěpení parkinsonského fenotypu. Po dobu více než dvaceti let se považovala za poměrně jasně definovanou poruchu s unikátní symptomatikou, která může být jen výjimečně předmětem diagnostického omylu. Poslední rok byly ale publikovány korelační ultrastrukturální studie, které nasvědčují tomu, že i drobné rozdíly v charakteristikách fenotypu kortikobazální degenerace mají patologický korelát; proto se nyní nemoc shrnuje pod označení kortikobazální syndromy.
|
|
|
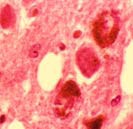
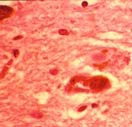
Histologické obrazy – demence s Lewyho tělísky Další fenotypy
Ještě složitější je situace s tzv. frontotemporální demencí. Toto onemocnění bylo známo více než 100 let jako Pickova demence; podle autora prvního popisu onemocnění Arnolda Picka, který se narodil ve Velkém Meziříčí. Pickova demence byla považována za jeden z typů presenilní demence, aniž se (po dobu celého století) uvažovalo o nějakém vztahu tohoto onemocnění k extrapyramidovému systému. Postupně se zjistilo, že Pickova nemoc je spíše konglomerátem různých typů presenilní demence a pro typickou lokalizaci změn dostal tento konglomerát název frontotemporální demence. Zahrnoval jak typy sporadické, tak i jasně dědičný typ frontotemporální demence s vazbou na chromozom 17.
Asi před deseti lety se začala častěji objevovat sdělení uvádějící, že někteří pacienti trpící frontotemporální demencí jeví i znaky parkinsonského fenotypu. Ještě později bylo jasné, že u řady těchto pacientů se nevyskytují jen příznaky parkinsonské, ale i příznaky amyotrofické laterální sklerózy, dalšího neurodegenerativního onemocnění, které však ve své původní podobě postihuje především míchu. To už začalo být hodně zajímavé – a to pro nápadnou podobnost tohoto syndromu s tím, čemu se říká „lytico-bodig“ v chamorrštině, a tzv. „guamský parkinsonský komplex“ v odborné literatuře. Jde o endemickou nemoc, vyskytující se pouze na ostrově Guam v Marianách a na poloostrově Kii ostrova Kjúšú v Japonsku. Poprvé ji popsali španělští misionáři před více než 200 lety (jedná se tedy o popis starší než Parkinsonův) a objevovala se na uvedených místech poměrně (až velmi) často až do 70. let minulého století, kdy její incidence začala pozvolna klesat.
Nemoc charakterizovala kombinace parkinsonismu s amyotrofickou laterální sklerózou a progredující demencí. Příčina nemoci byla dlouho neznámá. Nakonec se zjistilo, že ji způsobuje chronická intoxikace, jež vzniká požíváním masa malých netopýrů (tzv. Mariana fruit bat), kteří se téměř výlučně živí šťávou z cykasových plodů. Tato šťáva obsahuje specifický neurotoxin, který způsobuje pozvolné odumírání a zánik určitých nervových buněk.
Tím se dostáváme obloukem k osobě Johna Steela. Byl to právě on, kdo se se svým týmem dopátral celého potravního řetězce, jenž vede k rozvoji guamského parkinsonského komplexu. Jak velké tedy muselo být překvapení specialistů, kteří v posledních pěti letech opakovaně ve svých ordinacích v Evropě i Spojených státech viděli pacienty, kteří se až neuvěřitelně nápadně podobali těm z ostrova Guam – pacienty, kteří se několik let léčili pro klasickou Parkinsonovou nemoc a náhle se u nich objevily příznaky amyotrofické laterální sklerózy a progredující demence?
V první chvíli nebylo jasné, zda nejde o diagnostické omyly, ale skutečně se jednalo o pacienty, kteří nadále dobře odpovídali na léčbu L-DOPA, jejich demence dále progredovala a postupně přestali být schopni mluvit a polykat. Tedy jasný obraz guamského parkinsonského komplexu, o kterém však Steele poměrně přesvědčivě tvrdí, že má exotoxický původ. Takže trochu záhada. Vysvětlení dosud neexistuje a nemá smysl se pouštět do spekulací, neboť dat je zatím relativně málo a musíme pečlivě zkoumat všechny okolnosti, než vyslovíme alespoň hypotézu. Je však jasné, že existuje další parkinsonský fenotyp, který se pro nedostatek trefnější terminologie nazývá „neurodegenerative overlap syndrome“, tedy syndrom překrývající se neurodegenerace. Aspoň něco.
Výzkum pokračuje
Medicína je v zásadě biologická věda a jako ostatní biologické vědy se i ona rozvíjí na základě dalšího poznání ultrastrukturální morfologie a jejích souvislostí. I vývoj poznání parkinsonské neurodegenerace v posledních sto letech v podstatě refl ektuje zlepšené klinické pozorování déle přežívajících pacientů a rozmach morfologických a genetických metod.
Dnešní Parkinsonova nemoc je prostě jinou nemocí než nemoc, kterou kdysi popsal James Parkinson. Současný parkinsonský fenotyp je ze všeho nejvíce věrným odrazem progredující neurodegenerace. Ta může mít přímou, kaudokraniální progresi, kdy degenerativní proces postihuje postupně další a další část mozku ve směru od mozkového kmene k hemisférám. Potom se nemoc bude lékařům a okolí jevit jako velmi blízká Parkinsonovu popisu. Při jakékoli „odbočce“ z této relativně přímé cesty neurodegenerace se však rozvine jiný fenotyp než onen klasický – tedy některý z výše popsaných fenotypů, které pro nedostatek lepší terminologie nazýváme „atypickými“.
Bližší vztahy mezi klasickým obrazem onemocnění a obrazy atypickými a mezi charakterem a místem neurodegenerace zatím mapujeme; probíhá a zároveň i startuje řada výzkumných projektů, jejichž hlavním cílem je biologická charakterizace a klasifikace atypických parkinsonských syndromů. K bližšímu poznání těchto onemocnění a stanovení priorit dalšího výzkumu nepochybně přispívají i odborné konference podobně jako ta, již nyní 9. a 10. prosince pořádáme v Olomouci a na kterou jsou všichni specialisté z oborů neurologie, psychiatrie a patologie srdečně zváni.
|
|
|
|
|
 |
obsah čísla 84 |
 |
ročník 2010 |
|
témata |
|

| SANQUIS PLUS |

|
| GALERIE SANQUIS |

|
| PORADNA |

|
|