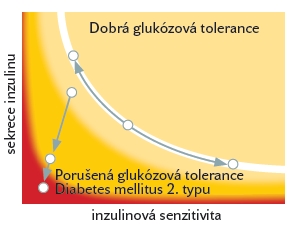
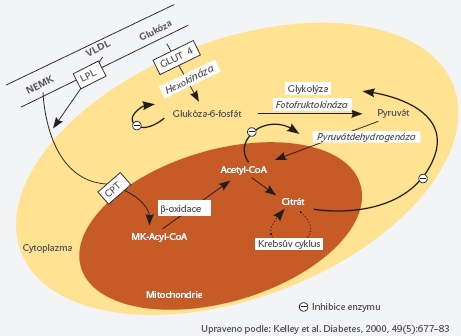
Metabolismus tukové tkáně v etiopatogenezi inzulinové rezistence Nadměrná akumulace tukových zásob v organismu je asociována se vznikem řady závažných onemocnění a patologických stavů, zejména s rozvojem inzulinové rezistence, diabetes mellitus 2. typu, hypertenze, aterosklerózy, dyslipidemie, syndromem polycystických ovarií, řadou nádorových onemocnění, artrózou nosných kloubů, dnou, psychologickými obtížemi atd. Obézní jedinci jsou často subjekty sociální diskriminace počínající již v dětství a projevující se později např. ve znevýhodnění při hledání zaměstnání či v dosažení nižšího stupně vzdělání. Z evolučního hlediska umožňovala tuková tkáň a v ní lokalizované adipocyty (buňky specializované na akumulaci energetických zásob získaných během relativního kalorického nadbytku) přežití v časech nedostatku nebo úplné absence potravy. Adipocyty ve vhodné kvantitě, lokalizaci a kvalitě jsou nepostradatelné a nezastupitelné i pro přežití současného Evropana, neboť ostatní tkáně lidského organismu nejsou uzpůsobeny pro ukládání lipidů. Pokud jsou ovšem zásobní schopnosti tukové tkáně překročeny (například u obézních pacientů), hypertrofované adipocyty již nejsou schopny akumulovat další množství lipidů a dochází k ukládání lipidů do jiných tkání, zejména jaterní a svalové. Takto ektopicky uložené lipidy závažným způsobem narušují metabolismus postižených buněk a tkání, způsobují poruchy v regulaci glukózového metabolismus a přispívají k dysfunkci a apoptóze (programované smrti) takto postižených buněk. Klinickým korelátem pak jsou chorobné stavy označované jako jaterní steatóza, non alkoholická steatohepatitida (NASH), inzulínová rezistence či diabetes mellitus 2. typu. Podrobné patofyziologické mechanismy a mediátory zprostředkovávající vazbu mezi nadměrnou kvantitou tukové tkáně na straně jedné a zvýšeným rizikem vzniku diabetes mellitus 2. typu na straně druhé jsou předmětem intenzivního studia v posledních několika desetiletích. Dlouho známou funkcí tukové tkáně je hydrolýza triglyceridů (složených z molekuly glycerolu a 3 molekul mastných kyselin) uložených v tukové kapénce uvnitř tukové buňky. Při tomto procesu dochází k uvolnění mastných kyselin do cirkulace, kde jsou označovány jako volné, ne-esterifikované mastné kyseliny (NEMK). O Jejich úloze v patogenezi inzulínové rezistence i pankreatické dysfunkce bude pojednáno v následujících odstavcích. V poslední době se věnuje značná pozornost i řadě působků proteinové povahy, které jsou produkovány a uvolňovány z tukové tkáně a mají úzký vztah k regulaci inzulinové senzitivity ve svalu, játrech a ovlivňují rovněž sekreci inzulínu v pankreatu. Tyto proteiny/hormony jsou označovány souhrnně jako „adipokiny“. Na jejich produkci se podílejí jak samotné tukové buňky, tak i ostatní buňky přítomné v tukové tkáni, z nichž jsou v tomto kontextu nejdůležitější makrofágy, které zvýšeně infiltrují tukovou tkáň v průběhu rozvoje obezity a pravděpodobně se podílejí na dysfunkci tukové tkáně provázející obezitu. Ne-esterifikované mastné kyseliny v rozvoji inzulínové rezistence Cirkulující ne-esterifikované mastné kyseliny (NEMK) mají řadu fyziologických funkcí. Jsou významným zdrojem energie zejména pro pracující sval, představují majoritní energetický substrát během hladovění a v průběhu těhotenství mají vliv při vzniku relativní inzulinorezistence matky a usnadňují tak dostupnost glukózy pro fétus. Vedle svého fyziologického významu mají NEMK pravděpodobně také význam v patogenezi inzulínové rezistence, jelikož obézní jedinci a pacienti s diabetes mellitus 2 typu mají obvykle vyšší hladinu NEMK v porovnání se zdravými subjekty. Elevace NEMK byla demonstrována jako nezávislý rizikový faktor progrese inzulínové rezistence a vzniku diabetes mellitus 2. typu. Efekt ne-esterifikovaných mastných kyselin se v kontextu porušené glukózové homeostázy nejvíce projevuje v interakci se svalovou, jaterní a endokrinní pankreatickou tkání. Mastné kyseliny a svalová tkáň Již v roce 1963 s revizí v roce 1998 popsal Randle vzájemnou kompetici mezi oxidativní utilizací glukózy a mastných kyselin a předložil první komplexní hypotézu o negativním působení NEMK na regulaci glykémie. Podle této teorie dochází při nadbytku mastných kyselin v cirkulaci k jejich preferenčnímu metabolismu ve svalové a jaterní tkáni. V průběhu degradace (oxidace) mastných kyselin dochází v buňce k produkci řady meziproduktů, zejména citrátu, acetyl-CoA (acetal koenzym-A) a NADH (nicotinamid adenin dinucleotid). Takto nahromaděné meziprodukty přímo inhibují metabolické dráhy a enzymy účastnící se nitrobuněčného metabolismu glukózy (pyruvát-dehydrogenáza, fosfofruktokináza). Konečným důsledkem komplexních změn je inhibice enzymu hexokináza, který fosforyluje molekulu glukózy po jejím vstupu do svalové buňky a představuje tak klíčový regulátor vstupu glukózy do svalové buňky. Při nižší aktivitě svalové hexokinázy se významně omezuje schopnost svalové buňky přijímat a utilizovat glukózu, vzniká inzulínová rezistence na úrovni svalové buňky. Schematicky je tento proces znázorněn na Obrázlku č. 2. Později byla tato hypotéza rozšířena o další komplementární mechanismy vycházející z poznatků molekulární biologie. Během energetického nadbytku a vyšší dostupnosti NEMK dochází ve svalové buňce rovněž k akumulaci meziproduktů oxidace mastných kyselin, zejména molekuly sestávající z řetězce mastné kyseliny navázané na acetyl-koenzym A (LCFA-CoA, long-chain fatty acid-coenzyme A). Tento meziprodukt inhibuje řadu dalších enzymů zapojených do utilizace a vychytávání glukózy jakož i do syntézy glykogenu. Navíc může být LCFA-CoA dále přeměněn na diacylglycerol (DAG), což je jedna z hlavních nitrobuněčných signalizačních molekul. Bylo opakovaně prokázáno, že DAG přímo interaguje a výrazně tlumí nitrobuněčnou signalizační kaskádu spouštěnou po navázání inzulínu na příslušný buněčný receptor. Rovněž přímá interakce nahromaděných mastných kyselin s glukózovým transportním proteinem (přenášejícím glukózu skrze buněčnou membránu do nitra svalové buňky) se může podílet na vzniku inzulínové rezistence. Těmito mechanismy je konečný účinek inzulínu na buňku metabolizující vyšší množství mastných kyselin snížen a buňka-tkáň-organismus se stává inzulíno-rezistentní. Mastné kyseliny a jaterní tkáň Rozvoje inzulínové rezistence v játrech je rovněž úzce spjat s působením ne-esterifikovaných mastných kyselin. U zdravých lidských probandů i u pacientů s diabetes mellitus 2. typu bylo prokázáno zvýšení produkce glukózy (glukoneogeneze) při experimentálním zvýšení hladiny volných mastných kyselin. U zdravých jedinců ovšem díky jaterní autoregulaci dochází paralelně se zvýšením glukoneogeneze k potlačení glykogenolýzy (degradace zásobního glykogenu) a celková produkce glukózy v játrech se po stimulaci NEMK nemění. Porucha této autoregulace u pacientů s diabetes mellitus 2.typu vede ke zvýšení glykogenolýzy i glukoneogeneze současně a jaterní tkáň se tak stává zdrojem glukózy uvolňované do cirkulace. Tato zvýšená nálož glukózy ovšem přichází za situace, kdy svalové buňky díky paralelně vzniklé inzulínové rezistenci nejsou schopny glukózu dostatečně z cirkulace odklízet. Má-li být za těchto okolností udržena normální hladina glukózy v krvi, musí být zvýšena produkce inzulínu v pankreatických β-buňkách. Mastné kyseliny a pankreatické β-buňky Ani pankreatické β-buňky nejsou uchráněny vlivům zprostředkovaným cirkulujícími ne-esterifikovanými mastnými kyselinami. Krátkodobá (< 6 hodin) expozice β-buněk NEMK vede ke zvýšení glukózou-stimulovaného výdeje inzulínu z β-buněk. Intenzita této stimulace je přitom přímo úměrně závislá na délce řetězce a stupni nenasycenosti mastné kyseliny. Přesné molekulární mechanismy zodpovědné za tento efekt nejsou do detailu stanoveny, nicméně podobně jako v případě svalové a jaterní tkáně se předpokládá alespoň částečný podíl LCFA-CoA. Přesně opačné účinky má ovšem dlouhodobé působení vyšší koncentrace NEMK, které vede ke snížení glukózou-stimulované sekrece inzulínu z pankreatu. Mezi molekulárními mechanismy zodpovědnými za tento jev je zvažována interakce NEMK s glukózovým transportérem, indukce oxidativního stresu a zvýšená intracelulární produkce ceramidu. Rovněž interakce NEMK s biosyntézou inzulínu, proinzulinu a transkripcí genu pro inzulín byly popsány a mohou se podílet na výše popsaném efektu. Na tomto místě je ovšem potřeba upozornit, že výše uvedená pozorování vlivu NEMK na pankreatický výdej inzulínu jsou založena na zvířecích modelech a in-vitro experimentech. Potvrzení těchto regulací in vivo v humánních experimentech je složitější, zejména z metodologického hlediska. Nicméně dosud provedené humánní studie potvrzují předchozí nálezy na modelech a tkáňových kulturách. Krátkodobá expozice vyšší hladině NEMK zvyšuje sekreci inzulínu přesně v takové míře, aby byl kompenzován pokles v periferní inzulínové senzitivitě. Při protrahované expozici NEMK se však tato regulace ztrácí a β-buňky nejsou schopny adekvátně zvýšit sekreci inzulínu, dochází tedy jak k poklesu periferní inzulínové senzitivity, tak k poklesu sekrece inzulínu. V takové situace je porušena regulace glykémie, rozvíjí se porucha glukózové tolerance a diabetes mellitus 2. typu. Intramyocelulárně uložené lipidy ve vztahu k inzulínové rezistenci Faktory, které mohou zhoršit inzulínovou senzitivitu svalové tkáně byly částečně uvedeny v předchozím textu. Vedle negativního působení volných mastných kyselin na metabolismus myocytů se v posledních letech diskutuje i úloha ektopicky uložených lipidů v cytoplazmě svalové buňky. Za situace, kdy hypertrofované adipocyty, jaké typicky nalézáme u obézních pacientů, již nejsou schopny ukládat potravou přijímané lipidy, dostávají se tyto do nitra jiných buněk, které nejsou k tomuto účelu uzpůsobené. V případě svalové buňky dochází při ektopické akumulaci lipidů k závažným poruchám v metabolismu svalu a rozvoji inzulínové rezistence. Nepříznivá situace může být dále umocněna nízkou pohybovou aktivitou daných jedinců. Za těchto okolností nejsou nahromaděné lipidy ve svalové buňce dostatečně metabolizovány k uhrazení energetických nároků svalové kontrakce (během fyzické zátěže) a inzulínová rezistence se prohlubuje. Molekulární mechanismy zodpovědné za vznik inzulínové rezistence ve svalové buňce v důsledku nahromadění lipidových inkluzí jsou podobné s mechanismy, kterými negativně působí mastné kyseliny, jež jsou popsané v předchozím textu. Klíčovou roli zde sehrává interakce s nitrobuněčnými signalizačními kaskádami, které jsou aktivovány po navázání inzulínu na membránový receptor. Schopnost myocytů efektivně metabolizovat lipidy je dána také počtem mitochondrií a aktivitou mitochondriálních enzymů. U obézních pacientů a diabetiků 2. typu byla demonstrována řada poruch snižujících oxidační kapacitu svalové buňky, například nižší aktivita klíčového enzymu zajišťujícího transport mastných kyselin do mitochondrií, CPT-1 (carnitin-palmitoyl transferáza) a nižší aktivita mitochondriálních enzymů zapojených v oxidačním řetězci. Pro klinickou praxi je důležitý poznatek, že vhodně vedený pohybový trénink vede u obézních jedinců a pacientů s diabetes mellitus 2. typu ke zvýšení počtu mitochondrií a větší oxidaci extra- i intra-celulárně uložených lipidů, čímž se zlepšuje citlivost svalové buňky k účinkům inzulínu. V souvislosti s množstvím a kvalitou kosterní svalové tkáně je velmi důležité zmínit přirozené ubývání kosterní svalové hmoty během stárnutí, které může dosáhnout až 25-40 % (mezi 25-60 rokem života). Rychlost úbytku svalové tkáně se rapidně zrychluje po 50. roce života a postihuje obě pohlaví. Z epidemiologických studií vyplývá, že prevalence sarkopenie dosahuje více než 50 % u osob starších 80 let. Vezmeme-li v potaz nezbytnost svalové tkáně pro inzulínem-stimulovaný odsun glukózy, nabízí se hypotéza, že věkem podmíněná sarkopenie může zhoršovat inzulínovou rezistenci a vést k manifestaci diabetes mellitus 2. typu. V současné době nejsou k dispozici jednoznačné důkazy, které by tuto hypotézu nezvratně prokázaly či vyvrátily. Z praktického hlediska je ovšem důležité upozornit na studie jednoznačně prokazující zlepšení inzulínové senzitivity po vytrvalostním a zejména silovém či silově‑dynamickém tréninku, bez ohledu na věk studovaných subjektů. Správně vedený silový trénink vede ke zvýšení svalové hmoty a pozorované zlepšení inzulínové rezistence je přítomno i bez redukce tukové tkáně. Tuková tkáň jako endokrinní orgán V posledních zhruba 15-ti letech byla identifikována řada látek proteinové povahy, které jsou produkovány tukovou tkání a následně uvolňovány buď do bezprostředního okolí buněk v tukové tkáni nebo do systémové cirkulace. Tyto produkty jsou souhrnně označovány jako „adipokiny“ a na jejich produkci se vedle samotných adipocytů podílejí velkou měrou i makrofágy a další imunokompetentní buňky lokalizované v tukové tkáni. Některé z adipokinů mají efekt pouze parakrinní a působí tudíž na adipocyty a další buňky přítomné v tukové tkáni (makrofágy, fibroblasty, endotelie, prekurzorové buňky). Většina adipokinů se ovšem dostává do krevního řečiště a působí pak typickým endokrinním způsobem ve vzdálených cílových orgánech (sval, játra, pankreas, mozek, endotelie). Na tukovou tkáň je v současné době nahlíženo jako na velmi potentní endokrinní tkáň regulující produkovanými hormony řadu dějů na úrovni celého organismu. Počet takto endokrinně aktivních molekul produkovaných v tukové tkáni dosahuje dle současných znalostí několika desítek, a jistě se bude i nadále zvyšovat. Spektrem svého účinku představují velmi heterogenní skupinu, zahrnující molekuly podílející se na regulaci intermediárního metabolismu, centrální regulaci příjmu potravy, tkáňové utilizaci substrátů i regulaci inzulínové senzitivity. Mnohé mají intimní vliv k etiopatogenezi aterosklerózy, jiné jsou zodpovědné za regulaci imunitních odpovědí a spolupodílejí se tak na vytváření mírného prozánětlivého stavu, který pravděpodobně přispívá k rozvoji inzulínové rezistence a kardiovaskulárních onemocnění. Obezita je charakterizována porušenými plazmatickými hladinami těchto cirkulujících adipokinů a rozvojem prozánětlivého stavu, který je asociován s rozvojem inzulínové rezistence a vznikem metabolických onemocnění jako jsou diabetes mellitus 2. typu a ateroskleróza. Podrobný popis všech adipokinů přesahuje rámec tohoto sdělení, proto budou uvedeny pouze tři zástupci této široké rodiny molekul: adiponectin, leptin a tumor necrosis factor-α. Adiponectin Adiponectin byl poprvé popsán v roce 1995. Jedná se o protein jehož produkce probíhá téměř výlučně v adipocytech. V plazmě je přítomen v překvapivě velkém množství (koncentrace činí 2-20 mg/l u zdravých jedinců, přibližně 0.01 % všech plazmatických proteinů). Adiponectin tvoří mezi ostatními adipokiny výjimku v tom smyslu, že jeho plazmatická hladina je snížena u osob s nadbytkem tukových zásob, diabetiků 2. typu a osob s ischemickou chorobou srdeční, zatímco koncentrace ostatních adipokinů v plazmě jsou u těchto stavů většinou zvýšeny. Adipocyty pravděpodobně za těchto patologických stavů nejsou schopny produkovat adiponectin v odpovídající míře, ovšem mechanismy zodpovědné za tuto poruchu nejsou dosud známé. Adiponectin významným způsobem zasahuje do regulace sacharidového a lipidového metabolizmu. Parenterální podání adiponectinu vede k výraznému zlepšení inzulínové rezistence na zvířecím modelu diabetu, dochází ke zvýšení utilizace a transportu glukózy a mastných kyselin ve svalových, jaterních i tukových buňkách. Zároveň adiponectin potlačuje glukoneogenezi v hepatocytech. Všechny tyto mechanismy přispívají k udržení normoglykémie. Dlouhodobá aplikace adiponectinu vede ke snížení hmotnosti u myší krmených vysokotukovou dietou, aniž by došlo ke snížení množství přijaté potravy. Tyto metabolické a inzulín-senzitizující účinky jsou pravděpodobně na molekulární úrovni zprostředkovány aktivací nitrobuněčného enzymu AMP-kináza (adenosin-monofosfát aktivovaná kináza), který představuje jakousi centrální buněčnou energetickou výhybku detekující nedostatek či nadbytek energie pro buněčné procesy. K aktivaci AMP-kinázy dochází při poklesu cytoplazmatické koncentrace hlavního energetického substrátu pro buněčné pochody - ATP (adenosin-trifosfátu). Aktivace AMP-kinázy vede v konečném důsledku ke zvýšení oxidace lipidů a glukózy v buňce a tím k obnovení intracelulárních hladin ATP a energetické rovnováhy v buňce. Ze studií provedených na lidských dobrovolnících vyplývá, že plazmatická koncentrace adiponectinu negativně koreluje s obsahem tuku v těle, BMI (body mass index), koncentrací triglyceridů, lačnou glykémií, lačnou hladinou inzulínu a různými parametry inzulínové rezistence. Naopak plazmatická hladina HDL cholesterolu je s hladinou adiponectinu asociována pozitivně. Někteří autoři dokládají, že adiponectin by mohl být vhodným markerem k odlišení obézních subjektů s dosud zachovalou citlivostí periferních tkání k účinkům inzulínu od obézních pacientů s již rozvinutou inzulínovou rezistencí. Existují rovněž nezanedbatelné pohlavní rozdíly v plazmatické hladině adiponectinu s vyšším obsahem adiponectinu u ženského pohlaví, adiponectin by se tak mohl spolupodílet na nižší incidenci kardiovaskulárních chorob u žen. Adiponectin se rovněž výrazně uplatňuje v ochraně arteriální stěny před vznikem a progresí aterosklerózy. Zabraňuje transformaci makrofágů v pěnité buňky a snižuje expresi povrchových adhesních molekul na povrchu makrofágů. Zároveň má adiponectin schopnost adherovat k obnaženému subendoteliálnímu prostoru poškozené cévy a významně omezovat nežádoucí proliferaci buněk hladkého svalstva a vaziva a tudíž hyperplázii medie během reparačních procesů. Leptin Leptin je dalším příslušníkem rodiny adipokinů, který je v menší míře produkován také v jiných tkáních (žaludek, játra, placenta, sval). Leptin má zásadní podíl v dlouhodobém udržování tělesné hmotnosti působením na hypothalamická centra regulující příjem potravy. Leptin zde inhibuje neurony zodpovědné za příjem potravy a naopak stimuluje aktivitu sympatického autonomního nervového systému a tak zvyšuje bazální energetický výdej. Se zvyšujícím se množstvím tukové tkáně stoupá plazmatická koncentrace leptinu a skrze jeho účinek na hypothalamická centra následně dochází zpětnovazebně k potlačení chuti k jídlu a zvýšení energetického výdeje. Leptin tak za fyziologických podmínek funguje jako dlouhodobý regulátor tukových zásob v organismu. Recentní práce poukazují i na vztah leptinu k hypothalamickým centrům regulujícím reprodukční funkce a sekreci gonadotropinů, kdy leptin funguje pravděpodobně jako indikátor dostatečných tukových (energetických) zásob nutných k pokrytí energetických nákladů reprodukce a těhotenství. U pacientek s patologicky nízkou hladinou leptinu (např. mentální anorexie) nalézáme typicky amenorrheu, která po normalizaci tukových zásob organismu ustupuje. Vrozený deficit leptinu či porucha funkce leptinového receptoru se u lidských subjektů fenotypicky manifestuje těžkou obezitou, dyslipidemií a inzulínovou rezistencí. Parenterálním podáváním uměle vyrobeného leptinu pacientům s poruchou v leptinovém systému jsou příznaky potlačeny a tělesná hmotnost normalizována. Je ovšem potřeba upozornit, že genové mutace v leptinovém systému manifestující se nízkou (nulovou) hladinou leptinu v plazmě nebo poruchou v leptinovém receptoru představují jednu z velmi vzácných forem tzv. monogenně podmíněné obezity. U naprosté většiny obézních pacientů však nalézáme naopak vysokou hladinu leptinu a terapeutické užití rekombinantního leptinu se ukázalo pro drtivou většinu obézních pacientů jako zcela neúčinné. Kromě účinků na centrální nervový systém působí leptin také v periferních metabolicky aktivních tkáních a pravděpodobně se tímto aditivním účinkem podílí na regulaci inzulínové senzitivity. V tukové tkáni leptin přímo interaguje jak s účinky inzulínu na buněčný metabolismus (glukózový transport, aktivace syntézy glykogenu, podpora ukládání lipidů, inhibice lipolýzy) tak s vazbou inzulínu na inzulínový receptor. Leptin negativním způsobem ovlivňuje pankreatické β-buňky, kde snížuje jejich schopnost produkovat inzulín v závislosti na stoupající glykémii. Významné jsou i účinky leptinu na jaterní buňky, kde se podílí na regulaci syntézy lipoproteinů. Tumor necrosis factor - α TNFαTNFα (tumor necrosis factor α, kachexin) byl historicky prvním z řady adipokinů, který byl navržen jako spojující článek mezi obezitou a inzulínovou rezistencí. Je produkován zejména buňkami imunitního systému lokalizovanými v tukové tkáni. Na zvířecím modelu a in vitro indukuje TNFα inzulínovou rezistenci přímou interakcí s inzulínovou signalizační kaskádou. TNFα také mohutně aktivuje lipolýzu v tukové tkáni, čímž dochází ke zvýšení hladiny volných mastných kyselin v plazmě. U obézních pacientů je exprese genu pro TNFα v podkožní tukové tkáni zvýšena a někteří autoři pozorovali pokles plazmatických hladin toto proteinu i jeho genové exprese v tukové tkáni po redukci hmotnosti. Další mechanismy podílející se na vzniku inzulínové rezistence Kromě uvedených procesů, které přispívají ke vzniku inzulínové rezistence a porušené produkci inzulínu v pankreatu u obézních pacientů, existují i další pochody, jenž se na vzniku a udržení tohoto stavu spolupodílejí. Jejich podrobný výčet přesahuje rámec tohoto sdělení, ovšem uvedeme alespoň některé z nich pro doplnění a dokreslení komplexnosti dané problematiky. Oxidační stres Metabolické zpracování sacharidů a lipidů probíhá v buňkách za dostatečného přístupu kyslíku v Krebsově cyklu. Tento proces se obecně nazývá oxidativní fosforylace a jejími konečnými produkty jsou oxid uhličitý, voda a energie vázaná do molekuly ATP. V průběhu Krebsova cyklu dochází k tvorbě molekuly NADH (nicotinamid-adenin-dinucleotid), která je nezbytná pro správný průběh cyklu. NADH je zodpovědný za přenos H+ iontů k vnitřní straně mitochondriální membrány, kde je využit pro syntézu ATP. Za běžných podmínek je NADH rychle využíván k syntéze ATP a nedochází k jeho hromadění v mitochondriích. Pokud jsou ovšem buněčné nároky na tvorbu ATP nízké a nabídka nutrientů nadměrná, není NADH dostatečně rychle degradován a v mitochondriích dochází k excesivnímu přenosu elektronů na atom kyslíku a tím k tvorbě vysoce reaktivních molekul tzv. „kyslíkových radikálů“. Mezi tyto molekuly jsou řazeny zejména superoxidové anionty (O2-), peroxid vodíku (H2O2) a hydroxylový radikál (.OH). Tyto agresivní molekuly mají negativní dopad na mnoho buněčných funkcí (poškozují strukturu DNA, lipidů a proteinů) a jejich zvýšené množství pravděpodobně přispívá k rozvoji řady onemocnění včetně inzulínové rezistence, aterosklerózy a nádorového bujení. Koncept oxidativního stresu byl úspěšně aplikován i v modelech etiopatogeneze pankreatické dysfunkce. Experimentálně bylo prokázáno in vitro i in vivo na lidských dobrovolnících, že zvýšená produkce kyslíkových radikálů zásadním způsobem snižuje první fázi sekrece inzulínu z pankreatu a podílí se tak na rozvoji pankreatické dysfunkce. Oxidační stres se může podílet na progresi od inzulínové rezistence přes porušenou glukózovou toleranci k manifestnímu diabetes mellitus 2. typu. Zvýšená hladina inzulínu, volných mastných kyselin i glukózy vede k porušení časné fáze sekrece inzulínu a rozvoji postprandiální hyperglykémie. Postupem času dochází dalším působením kyslíkových radikálů k ireverzibilnímu poškození β-buněk a jejich postupnému zániku, rozvíjí se manifestní diabetes. Stres endoplazmatického retikula Endoplazmatické retikulum (ER) je buněčná organela přítomná v eukaryotických buňkách složená z rozsáhlého systému tubulů, váčků a cisteren. V ER kde probíhá řada specializovaných procesů, zejména syntéza proteinů, jejich formování do konečné prostorové struktury a následný transport do buněčné membrány či jejich sekrece do mimobuněčného prostoru. Probíhá zde i syntéza a uskladnění glykogenu, steroidů a dalších látek. Při nadměrně zvýšené dostupnosti živin dochází k funkčnímu zatížení tohoto systému a k vyvolání specifického stavu označovaného jako stres endoplazmatického retikula s aktivací řady signalizačních drah, jejichž cílem je pomocí změn v buněčných pochodech a genové expresi normalizovat mikroprostředí v ER a udržet tak buněčnou integritu. Součástí této komplexní buněčné reakce je také omezení inzulínové buněčné signalizace a vyvolání stavu inzulínové rezistence dané buňky. Navržená hypotéza staví tedy ER do pozice styčného bodu mezi metabolickými změnami a aktivací signálních drah spojených se zánětlivou signalizací. Při experimentálních zásazích do buněčných odpovědí vyvolaných stresem endoplazmatického retikula dochází k výrazným patologickým projevům zejména v buňkách s vysokou sekreční aktivitou, mezi které patří typicky např. pankreatické β-buňky. Platnost tohoto mechanismu u lidských subjektů je ovšem ještě potřeba ověřit. Obrázek 2. Schematické znázornění metabolické interakce mezi mastnými kyselinami a sacharidy v buňce - Randlův cyklus
|