PRION DISEASES AND THE IMMUNE SYSTEM Mgr. Hana Glierová, Dr.Ing. Karel Holada
Ústav imunologie a mikrobiologie, 1.LF UK, Praha Souhrn
Prionové choroby, také nazývané transmisivní spongiformní encefalopatie (TSE), jsou smrtelná neurodegenerativní onemocnění postihujících člověka i zvířata. V průběhu prionového onemocnění dochází k ukládání abnormálního prionového proteinu (PrPtse, prion) ve formě amyloidních plaků v centrálním nervovém systému (CNS), který je jediným funkčně postiženým orgánem. Po infekci priony dochází nejprve k akumulaci PrPtse v lymfatické tkáni a následně k invazi do CNS. Hlavní úlohu v periferní replikaci prionů hrají folikulární dendritické buňky (FDC), které jsou rovněž nezbytné pro neuroinvazi. Během TSE infekce dochází k upregulaci některých složek komplementu, nicméně imunitní systém nereaguje tvorbou protilátek, dokonce se podílí na šíření prionů v organismu. Poznatky o úloze imunitního systému v patogenezi prionových chorob mohou přispět k vývoji diagnostického testu pro detekci prionů v krvi a k nalezení účinné terapie. Mezi lidské prionové choroby patří Creutzfeldt-Jakobova choroba (CJD), Kuru, Gerstmann-Straussler-Scheinker syndrom (GSS) a fatální familiární insomnie (FFI). Prionové choroby postihující zvířata zahrnují například „scrapie“ ovcí, bovinní spongiformní encefalopatii (BSE) hovězího dobytka a „chronic wasting disease“ jelenovité zvěře (CWD).
Nejčastějším prionovým onemocněním TSE u lidí je CJD. Vyskytuje se celosvětově s frekvencí jedno až dvě úmrtí na milion obyvatel a rok. Přibližně 85 % případů vzniká sporadicky bez zjevné příčiny (sCJD), 10 až 15 % případů je podmíněno geneticky (familial, fCJD), přibližně 2 až 3 % připadají na iatrogenní přenos (transplantace rohovky či dura mater od infikovaného dárce, kontaminované růstové hormony, neurochirurgické nástroje, a v případě vCJD i krevní transfuze). V roce 1996 se ve Velké Británii objevila nová forma CJD - variantní CJD (vCJD) - zoonóza odvozená od BSE. Molekulární podstata prionových chorob
Klíčovým dějem v patogenezi prionových chorob je přeměna normálního buněčného prionového proteinu (PrPc) na abnormální, infekční formu (PrPtse, prion), která se od PrPc liší pouze konformací. Podle prionové teorie je infekční částice tvořena pouze proteinem bez účasti specifické nukleové kyseliny [1]. PrPc je membránový glykoprotein exprimovaný na mnoha tělních buňkách, jeho funkce však zatím není objasněna. Předpokládá se, že k propagaci PrPtse v buňce dochází přímým kontaktem s normálním PrPc, kterému vnutí svoji patologickou konformaci. Procesu konverze PrPc na PrPtse se pravděpodobně účastní i další dosud neznámé molekuly, např. tzv. faktor X [2].
Existují různé kmeny prionů („strains“) s rozdílnou konformací PrPtse, které se liší inkubační dobou, klinickými příznaky, délkou trvání onemocnění, vakuolizací CNS a tvorbou amyloidních plaků [3]. Prionové kmeny hrají roli v druhové specifitě prionů, např. přenos „scrapie“ z ovcí na člověka nebyl dosud zaznamenán, naproti tomu BSE priony jsou relativně druhově nespecifické a na člověka byly přeneseny ve formě vCJD.
Význam exprese PrPc na buňkách imunitního systému
PrPc je exprimován na řadě buněk imunitního systému, včetně leukocytů a folikulárních dendritických buněk (FDC), které exprimují vysoké hladiny PrPc. Exprese PrPc je regulována během vývoje lymfocytů v kostní dřeni a thymu a během následné mitogenní aktivace [4] . Existují značné mezidruhové rozdíly v expresi PrPc na leukocytech v periferii [5], které komplikují spekulace o fyziologické funkci PrPc.
Role imunitního systému v šíření prionů v organismu
Po orální infekci priony se nejprve infekční agens akumuluje ve folikulárních dendritických buňkách (FDC) v Peyerových placích v lymfatické tkáni asociované se střevy (GALT, „gut associated lymphoid tissue“) a v gangliích enterického nervového systému. Transport infekčních prionů z apikálního na bazolaterární povrch střevního epitelu není zcela objasněn, roli prostředníka mohou hrát M buňky. V mechanismu přenosu prionů ze střevního lumen do GALT mohou podle posledních poznatků mít významnou roli migrující hematopoetické CD11c+ dendritické buňky. Jejich deplece v GALT a slezině před orální inokulací priony u myší zabránila akumulaci prionů v těchto tkáních a snížila vnímavost k infekci [6]. Po replikaci v Peyerových placích a mesenteriálních lymfatických uzlinách priony přecházejí do lymfy a do krve a jsou transportovány do vzdálenějších lymfatických orgánů. Následuje pomnožení ve slezině, apendixu, mandlích a dalších orgánech, které však nevede, na rozdíl od mozku, k jejich funkční poruše. Po té dochází k infekci periferních nervů a invazi prionů do CNS.
Neuroinvaze je závislá na expresi PrPc v periferních nervech, které spojují lymfatické tkáně a CNS. Rovněž struktura lymfatických tkání zásadně ovlivňuje proces neuroinvaze. Na myším modelu bylo demonstrováno, že ablace receptoru pro chemokin 5 způsobující přiblížení FDC a hlavních slezinných nervů, vede k urychlení transportu intraperitoneálně aplikovaných prionů do míchy. Priony se s největší pravděpodobností šíří do CNS prostřednictvím enterického nervového systému nebo splanchnickými či vagovými nervy. V CNS se abnormální prionový protein akumuluje jak intracelulárně, tak extracelulárně ve formě amyloidních plaků.
Úloha imunitního systému v TSE
Úloha jednotlivých složek imunitního systému není zcela objasněna.
V průběhu prionových chorob dochází v CNS k rozsáhlé aktivaci a proliferaci mikrogliálních buněk, CNS rovněž infiltrují T-buňky. Během prionových onemocnění je zvýšena exprese prozánětlivých a efektorových cytokinů. Bylo prokázáno, že intracerebrální inokulace prionů vyvolala TSE u myší s různými imunodeficiencemi, jako je zejména deficience T-buněk, B-buněk, receptorů pro interferon, složek komplementu, TNFα, lymfotoxinů a receptorů pro chemokiny. Ve všech modelech po intracerebrální inokulaci probíhala patogeneze se stejnou kinetikou jako u imunokompetentních myší. To nasvědčuje tomu, že pokud priony proniknou do mozku, nemají již imunologické reakce na onemocnění vliv.
Úloha T- a B-lymfocytů v patogenezi TSE
T-lymfocyty se zřejmě nepodílí na patogenezi prionových chorob. Dokladem jsou výsledky studie ukazující, že thymektomie nemá vliv na inkubační dobu onemocnění vyvolaného periferní infekcí. Pozdější studie na transgenních a imunodeficientních myších potvrdily, že defekty v T-lymfocytech (CD4-/-, CD8-/-, β2-µ-/-, TCRα-/- a Perforin-/- myši) neměly vliv na vnímavost k onemocnění nebo akumulaci TSE infektivity ve slezině.
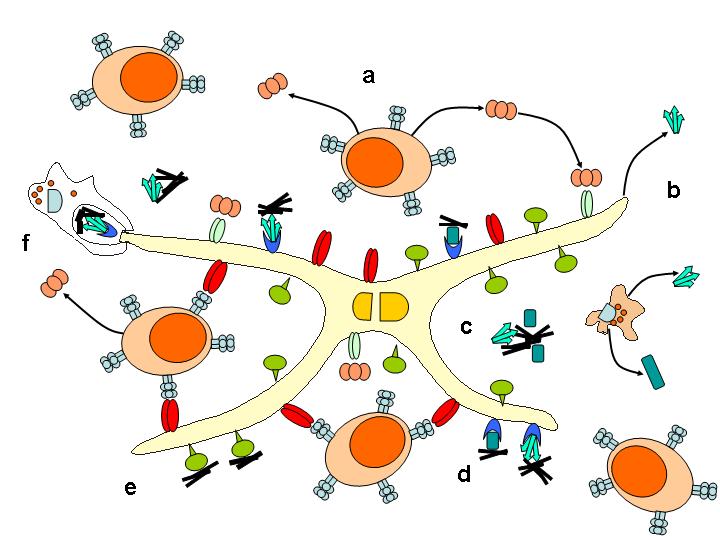
Oproti tomu B-lymfocyty hrají klíčovou roli v periferní patogenezi prionových chorob. U myší s deficitem B-lymfocytů je signifikantně narušená akumulace prionů ve slezině a následná neuroinvaze. Nicméně přítomnost PrPc na B-lymfocytech není pro proces neuroinvaze nezbytná. Většina B-lymfocytů se nachází ve slezině, v Peyerových placích či v lymfatických uzlinách. B-lymfocyty produkují tumor necrosis factor-α (TNF-α) a lymphotoxin α1β2, nezbytné pro dozrávání FDC v lymfoidních orgánech. Myši s deficitem B-lymfocytů zcela postrádají zralé FDC, které jsou nezbytné pro akumulaci prionů a jejich následnou neuroinvazi.
Úloha FDC v patogenezi TSE
Po periferní expozici infekčnímu agens je u většiny TSE neuroinvaze zásadně závislá na FDC. Akumulace a replikace prionů ve slezině vyžaduje zralé FDC. V jejich nepřítomnosti nedochází k akumulaci infekčního agens v lymfatické tkáni, neuroinvaze je zpomalena a je snížena vnímavost k infekci. FDC jsou schopné vázat na svém povrchu antigeny ve formě komplexů s protilátkou a/nebo složkami komplementu a syntetizovat C1q složku komplementu (Obr; převzato – ref [12]). Výsledky nedávné studie ukazují, že FDC jsou nezbytné pro vývoj onemocnění během krátké doby po perorální inokulaci. Při depleci FDC během tohoto období nedojde u myší k rozvoji onemocnění. Infektivita a akumulace PrPtse byly detekovány v přímé souvislosti s FDC v lymfatické tkáni pacientů s vCJD, ale ne u pacientů se sporadickou a iatrogenní CJD. FDC jako klíčové buňky v periferní patogenezi TSE mohou být cílem pro terapii v období mezi expozicí infekci a neuroinvazí.
Úloha makrofágů v patogenezi TSE
Makrofágy jsou podobně jako FDC dlouho žíjící buňky exprimují PrPc. Ve slezinných makrofázích myší infikovaných priony byla popsána intralysozomální akumulace prionů. Odstranění makrofágů před nebo krátce po periferní inokulaci priony vedlo ke zvýšenému hromadění PrPtse ve slezině a ke zkrácení inkubační doby onemocnění.
Úloha komplementu v patogenezi TSE
Nepřítomnost C1q, C2 a C3 složek komplementu nebo receptoru CD21/CD35, jehož prostřednictvím FDC vážou opsonizované antigeny, brání hromadění prionů ve slezině a prodlužuje inkubační dobu [7]. C1q a C3 jsou důležité pro akumulaci prionů ve FDC během prvních dnů po infekci [8]. Tato data naznačují, že priony jsou opsonizovány složkami komplementu C1q, C2 a C3. Opsonizace prionům umožňuje využít receptor pro komplement na povrchu FDC a usnadnit zakotvení a replikaci PrPtse na těchto buňkách. Mechanismus interakce PrPtse se složkami komplementu zatím není znám. Podle nejnovějších poznatků C1q může vázat PrPtse [9]. TSE agens zřejmě může vázat komplement buď přímo v místě expozice, nebo až na FDC. Komplementový systém by tak mohl umožňovat vychytávání prionů pohyblivými buňkami, jako jsou makrofágy nebo cirkulující dendritické buňky, které by se tak mohly podílet na transportu prionů z a do lymfatické tkáně.
Úloha komplementu v neurodegeneraci CNS
Gliové buňky a neurony mohou syntetizovat molekuly komplementu jako odpověď na infekci CNS. Aktivace komplementu představuje základní imunitní obranný mechanismus, ovšem jeho nekontrolovaná aktivace může způsobit neuropatologické změny; např. Alzheimerova choroba, Huntingtonova choroba a roztroušená skleróza jsou spojeny s chronickou aktivací a syntézou komplementu.
C1q a C3 složky komplementu byly detekovány v amyloidních placích v mozcích pacientů s variantní, sporadickou a familiární CJD a pacientů s GSS. Analýza genové exprese v mozcích hlodavců experimentálně infikovaných „scrapie“ nebo BSE ukazuje zvýšenou expresi C1q specifické mRNA [10].
Složky komplementu C5, C6, C7, C8, a C9 se účastní tvorby membránu atakujícího komplexu (MAC). Tento komplex vytváří póry v membráně cílových buněk, což vede k jejich lýze a buněčné smrti. MAC byl nalezen v mozcích jedinců infikovaných TSE, přičemž přítomnost MAC korelovala se závažností neurodegenerace. U jedinců infikovaných TSE byla rovněž pozorována zvýšená exprese clusterinu. Clusterin (apolipoprotein J) je glykoprotein, který mimo jiné inhibuje komplementem zprostředkovanou cytolýzu [11]. V průběhu TSEje clusterin asociován s deposity abnormálního prionového proteinu v CNS. Data získaná na experimentálně infikovaných hlodavcích naznačují, že exprese clusterinu je indukována hromaděním patologického prionového proteinu [11].
Závěr
Variantní CJD je jedinou lidskou TSE, u které je infekční prionový protein detekován ve větší míře mimo CNS. U pacientů s vCJD je PrPtse pravidelně detekován v lymfatické tkáni, v mandlích, apendixu a dalších orgánech lymfatického systému. Od roku 1996, kdy byla vCJD poprvé popsána, bylo ve Velké Británii zaznamenáno 163 případů tohoto onemocnění, 160 pacientů zemřelo (UK CJD Surveillance Unit, data k únoru 2008). V posledních letech byly zaznamenány čtyři případy přenosu vCJD krevní transfuzí. Zatím není k dispozici diagnostický test detekující priony v krvi. Poznatky o akumulaci PrPtse v lidské lymfatické tkáni mohou přispět nejen k vývoji diagnostického testu a stanovení možného rozsahu vCJD epidemie, ale i k vývoji chybějích terapeutických postupů. Poděkování
Podporováno granty GAČR 310/08/0878, GAČR 310/05/H533, VZ 20610027.
Kontaktní adresa
Dr. Ing. Karel Holada
Prionová laboratoř
Ústav imunologie a mikrobiologie
1. lékařská fakulta Univerzity Karlovy v Praze
Studničkova 7, 12800 Praha 2
email: karel.holada@lf1.cuni.cz Literatura
1. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 9;216(4542):136-44.
2. Cohen FE, Prusiner SB. Parhologic conformations of prion proteins. Annual Review of Biochemisrty 1998; vol.67:793-819.
3. Aguzzi A, Sigurdson ChJ. Antiprion immunotherapy: To supress or to stimulate? Nature 2004; vol.4: 725-736.
4. Mabbott NA, Brown KL, Manson J, Bruce ME. T-lymphocyte activation and the cellular form of the prion protein. Immunology 1997;92:161-5.
5. Holada K, Vostal JG. Different levels of prion protein (PrPc) expression on hamster, mouse and human blood cells. Br J Haem 2000; 110: 472-480.
6. Raymond CR, Aucouturier P, Mabbott NA. In vivo depletion of CD11c+ cells impairs scrapie agent neuroinvasion from the intestine. J Immunol 2007; vol.179:7758-7766.
7. Klein MA, Kaeser PS, Schwarz P, Weyd H, Xenarios I, Zinkernagel RM, Carroll MC, Verbeek JS, Botto M, Walport MJ et al. Complement facilitates early prion pathogenesis. Nat Med 2001, 7:488-492.
8. Mabbott NA. The complement system in prion diseases. Current opinion in immunology 2004; 16: 587-593.
9. Blanquet-Grossard F, Thielens NM, Vendrely C, Jamin M, Arlaud CJ. Complement protein C1q recognizes conformationally modified form of the prion protein. Biochemistry 2005; 44: 4349-4356.
10. Dandoy-Dron F, Benboudjema L, Guillo F, Jaegly A, Jasmin C, Dormont D, Tovey MG, Dron M: Enhanced levels of scrapie responsive gene mRNA in BSE-infected mouse brain. Molec Brain Res 2000; 76:173-17
11. Sasaki K, Doh-ura K, Ironside JW, Mabbott N, Iwaki T. Clusterin expression in follicular dendritic cells associated with prion protein accumulation. J Pathol 2006; 209: 484-491.
12. Mabbott NA, MacPherson GG. Prions and their lethal journey to the brain Nature Reviews in Microbiology 2006; 4:201-211.
|