|
reklama

| Obezita v etiopatogenezi inzulinové rezistence a diabetes mellitus 2. typu |
|
MUDr. Jan Polák Ph.D.
as. MUDr. Jan Brož |
| SANQUIS č.59/2008, str. 61 |
 |
zpět na výběr odborných článků |
| Ač bylo onemocnění charakterizované nadměrnou tvorbou sladké moči popsáno již v sanskrtském textu Atharvaveda (1500–1000 př. n. l.), je poznání mechanismů vedoucích k rozvoji diabetes mellitus 2. typu i v 21. století výzvou pro kliniky a vědce.
|
|
Komplexnost a heterogenita procesů vedoucích k poruše glukózového a lipidového metabolismu a interakce mezi genetickými faktory a vlivy prostředí jsou jen některé z aspektů, které významnou měrou komplikují výzkum v této oblasti. Ačkoli dosud není jednoznačně definován proces vzniku diabetes mellitus 2. typu, byla formulována řada hypotéz a identifikováno několik genetických determinant, které se pravděpodobně na vzniku této choroby větší či menší měrou podílejí.
V následujícím textu jsme se pokusili o zprostředkování současného pohledu na podstatu etiopatogeneze diabetes mellitus 2. typu. Některé z popisovaných mechanismů jsou detailně prozkoumány a je jim proto věnována větší pozornost, snažili jsme se však uvést i takové, jejichž zkoumání je teprve v počátcích.
Inzulinová rezistence, pankreatická dysfunkce a hladina glukózy v krvi
Zjednodušeně si lze představit, že aktuální hodnota glykémie je odrazem rovnováhy mezi příjmem sacharidů a vlastní produkcí glukózy organismem (zejména jaterní glukoneogenezí a glykogenolýzou) na straně jedné a na druhé straně její spotřebou jako energetického substrátu zejména v kosterní svalové tkáni či využitím k syntéze zásobních molekul (glykogenu či tukových zásob). Kosterní svalová tkáň, která zodpovídá za odsun 60–80 % glukózy po jejím příjmu, spolu s jaterní tkání, která naopak glukózu do cirkulace dodává, se výrazně podílejí na udržení glukózové homeostázy. Za fyziologických podmínek in-vivo je ovšem regulace glykémie značně složitější, důležitou roli sehrává dynamika vstřebávání sacharidů z gastrointestinálního traktu (GIT), produkce GIT hormonů s účinky na metabolismus glukózy (tzv. inkretiny) a uplatňují se i klasické glukoregulační hormony (kortisol, růstový hormon, thyroxin, glukagon).
Řadou experimentů bylo prokázáno, že poruchy v metabolismu jaterní a svalové buňky přímo ovlivňují celotělovou glukózovou homeostázu. Inzulin, základní hormonální regulátor v glukózovém metabolismu, zabezpečuje svými účinky na kosterní svalovou, jaterní a tukovou tkáň odsun glukózy z cirkulace. Citlivost těchto tkání k účinkům inzulinu je proto zásadním faktorem determinujícím míru odsunu glukózy z cirkulace.
Stav organismu, kdy cílové tkáně nejsou schopny adekvátně reagovat na inzulin, označujeme pojmem inzulinová rezistence. Za tohoto stavu je pro vyvolání očekávané odpovědi a tím k zachování normoglykémie nutné větší množství inzulinu, čímž se překoná přítomná inzulinová rezistence. Je nepochybné, že inzulinová rezistence je nezbytným faktorem v rozvoji diabetes mellitus 2. typu. Pro klinickou manifestaci tohoto onemocnění je ovšem nezbytné, aby byla současně přítomna také neschopnost pankreatických -buněk adekvátně zvyšovat sekreci inzulinu k udržení normální hladiny glykémie, tedy stav označovaný jako pankreatická dysfunkce.
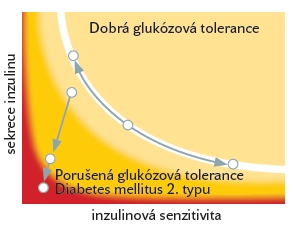
Z experimentálních i observačních studií vyplývá, že sekrece inzulinu z pankreatu v závislosti na rostoucí inzulinové rezistenci odpovídá hyperbolickému vztahu, viz. obr 1. Z tohoto grafu je patrné, že u pacientů s inzulinovou rezistencí dochází ke kompenzatornímu zvýšení sekrece inzulinu přesně v takové míře, aby byla udržena normální hladina glukózy v krvi. Vznik pankreatické dysfunkce znamená odklon od zobrazené křivky směrem dolů. Množství inzulinu uvolňovaného z pankreatických ostrůvků není dostatečné ke kompenzaci inzulinové rezistence a dochází zákonitě k porušení glukózové tolerance a rozvoji diabetes mellitus 2. typu. Obrázek 1 rovněž poskytuje fyziologický podklad tradiční terapie diabetes mellitus 2. typu. Docílit zlepšené glukózové tolerance lze u daného jedince zvýšením hladiny inzulinu (podáním zevního inzulinu parenterálně či zvýšením sekrece vlastního inzulinu deriváty sulfonylurey) či zlepšením inzulinové senzitivity periferních tkání (pohybovou aktivitou nebo metforminem či thiazolidindiony).
Obrázek č. 1 Hyperbolická závislost sekrece inzulínu na inzulínové senzitivitě
|
|
|
Metabolismus tukové tkáně v etiopatogenezi inzulinové rezistence Nadměrná akumulace tukových zásob v organismu je asociována se vznikem řady závažných onemocnění a patologických stavů, zejména s rozvojem inzulinové rezistence, diabetes mellitus 2. typu, hypertenze, aterosklerózy, dyslipidemie, syndromem polycystických ovarií, řadou nádorových onemocnění, artrózou nosných kloubů, dnou, psychologickými obtížemi atd. Obézní jedinci jsou často subjekty sociální diskriminace počínající již v dětství a projevující se později např. ve znevýhodnění při hledání zaměstnání či v dosažení nižšího stupně vzdělání.
Z evolučního hlediska umožňovala tuková tkáň přežití v časech nedostatku nebo úplné absence potravy. Pokud jsou ovšem zásobní schopnosti tukové tkáně překročeny (například u obézních pacientů), hypertrofované adipocyty již nejsou schopny akumulovat další lipidy a dochází k jejich uložení do jiných tkání, zejména jaterní a svalové. Takto ektopicky uložené lipidy závažným způsobem narušují metabolismus postižených buněk a tkání, způsobují poruchy v regulaci glukózového metabolismu a přispívají k dysfunkci a apoptóze (programované smrti) těchto buněk. Klinickým korelátem pak jsou chorobné stavy označované jako jaterní steatóza, non alkoholická steatohepatitida (NASH), inzulinová rezistence či diabetes mellitus 2. typu.
Podrobné patofyziologické mechanismy a mediátory zprostředkovávající vazbu mezi nadměrnou kvantitou tukové tkáně na straně jedné a zvýšeným rizikem vzniku diabetes mellitus 2. typu na straně druhé jsou předmětem intenzivního studia v posledních desetiletích. Dlouho známou funkcí tukové tkáně je hydrolýza triglyceridů uložených v tukové kapénce uvnitř tukové buňky. Při tomto procesu dochází k uvolnění mastných kyselin do cirkulace, kde jsou označovány jako volné, ne-esterifikované mastné kyseliny (NEMK). O jejich úloze v patogenezi inzulinové rezistence i pankreatické dysfunkce bude pojednáno v následujících odstavcích. V poslední době se věnuje značná pozornost i řadě působků proteinové povahy, které jsou produkovány a uvolňovány z tukové tkáně a mají úzký vztah k regulaci inzulinové senzitivity ve svalu, játrech a ovlivňují rovněž sekreci inzulinu v pankreatu. Tyto proteiny/hormony jsou označovány souhrnně jako „adipokiny“ a bude o nich rovněž pojednáno.
Ne-esterifikované mastné kyseliny v rozvoji inzulinové rezistence
Cirkulující ne-esterifikované mastné kyseliny (NEMK) mají řadu fyziologických funkcí. Jsou významným zdrojem energie pro pracující sval a představují majoritní energetický substrát během hladovění. Vedle svého fyziologického významu mají NEMK také význam v patogenezi inzulinové rezistence. Obézní jedinci a pacienti s diabetes mellitus 2. typu mají obvykle vyšší plazmatickou hladinu NEMK v porovnání se zdravými subjekty. Efekt ne-esterifikovaných mastných kyselin se v kontextu porušené glukózové homeostázy nejvíce projevuje v interakci se svalovou, jaterní a endokrinní pankreatickou tkání.
Mastné kyseliny a svalová tkáň
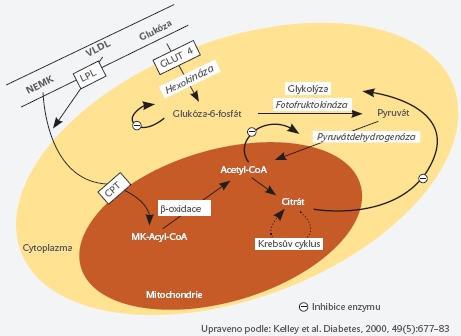
Již v roce 1963 popsal Randle vzájemnou kompetici mezi oxidativní utilizací glukózy a mastných kyselin ve svalu a předložil první komplexní hypotézu o negativním působení NEMK na regulaci glykémie. Podle této teorie dochází při nadbytku mastných kyselin v cirkulaci k jejich preferenčnímu metabolismu ve svalové a jaterní tkáni. V průběhu degradace (oxidace) mastných kyselin dochází v buňce k produkci řady meziproduktů, zejména citrátu, acetyl-CoA (acetyl koenzym-A) a NADH (nicotinamid adenin dinucleotid). Takto nahromaděné meziprodukty přímo inhibují metabolické dráhy a enzymy účastnící se nitrobuněčného metabolismu glukózy (pyruvát-dehydrogenáza, fosfofruktokináza). Konečným důsledkem komplexních změn je inhibice enzymu hexokináza, který fosforyluje molekulu glukózy po jejím vstupu do svalové buňky a představuje tak klíčový regulátor vstupu glukózy do svalové buňky. Při nižší aktivitě svalové hexokinázy se významně omezuje schopnost svalové buňky přijímat a utilizovat glukózu, vzniká inzulinová rezistence na úrovni svalové buňky (viz obrázek č. 2).
Během energetického nadbytku a vyšší dostupnosti NEMK dochází ve svalové buňce rovněž k akumulaci meziproduktů oxidace mastných kyselin, zejména molekuly obsahující řetězec mastné kyseliny navázané na acetyl-koenzym A (LCFACoA, long-chain fatty acid-coenzyme A). Tento meziprodukt inhibuje řadu dalších enzymů zapojených do utilizace a vychytávání glukózy, jakož i do syntézy glykogenu. Navíc může být LCFA-CoA dále přeměněn na diacylglycerol (DAG), což je jedna z hlavních nitrobuněčných signalizačních molekul. Bylo opakovaně prokázáno, že DAG přímo interaguje a výrazně tlumí nitrobuněčnou signalizační kaskádu spouštěnou po navázání inzulinu na příslušný buněčný receptor. Rovněž přímá interakce nahromaděných mastných kyselin s membránovým glukózovým transportním proteinem (GLUT-4, přenášejícím glukózu skrze buněčnou membránu do nitra svalové buňky) se může podílet na vzniku inzulinové rezistence. Uvedenými mechanismy je účinek inzulinu na danou buňku metabolizující vyšší množství mastných kyselin snížen a buňka-tkáň-organismus se stává rezistentní k inzulinu.
Obrázek 2. Schematické znázornění metabolické interakce mezi mastnými kyselinami a sacharidy v buňce - Randlův cyklus
|
|
|
Mastné kyseliny a jaterní tkáň
Rozvoj inzulinové rezistence v játrech je rovněž úzce spjat s působením NEMK. U zdravých lidských probandů i u pacientů s diabetes mellitus 2. typu bylo prokázáno zvýšení produkce glukózy (glukoneogeneze) při experimentálním zvýšení hladiny volných mastných kyselin. U pacientů s diabetes mellitus 2. typu dochází tak ke zvýšení glykogenolýzy i glukoneogeneze současně a jaterní tkáň se stává zdrojem glukózy uvolňované do cirkulace. Tato zvýšená nálož glukózy ovšem přichází za situace, kdy svalové buňky kvůli paralelně vzniklé inzulinové rezistenci nejsou schopny glukózu dostatečně z cirkulace odklízet. Má-li být za těchto okolností udržena normální hladina glukózy v krvi, musí být zvýšena produkce inzulinu v pankreatických β-buňkách. Mastné kyseliny a pankreatické β-buňky
Ani pankreatické β-buňky nejsou uchráněny před účinkem cirkulujících NEMK. Dlouhodobé působení vyšší koncentrace NEMK vede ke snížení glukózou stimulované sekrece inzulinu z pankreatu. Mezi molekulárními mechanismy zodpovědnými za tento jev je zvažována interakce NEMK s glukózovým transportérem, indukce oxidativního stresu a zvýšená intracelulární produkce ceramidu. Rovněž interakce NEMK s biosyntézou inzulinu, proinzulinu a transkripcí genu pro inzulin byly popsány a mohou se podílet na výše popsaném efektu.
Při protrahované expozici NEMK ztrácejí β-buňky schopnost adekvátně zvýšit sekreci inzulinu a dochází ke vzniku pankreatické dysfunkce. Pokud je zároveň přítomna inzulinová rezistence ve svalové tkáni a produkce glukózy v játrech, dochází k porušení glukózové homeostázy a vzniku diabetes mellitus 2. typu.
Intramyocelulárně uložené lipidy ve vztahu k inzulinové rezistenci
Kromě negativního působení volných mastných kyselin na metabolismus myocytů se v posledních letech diskutuje i úloha ektopicky uložených lipidů v cytoplazmě svalové buňky. V případě svalové buňky dochází při ektopické akumulaci lipidů k závažným poruchám v metabolismu svalu a rozvoji inzulinové rezistence. Molekulární mechanismy zodpovědné za vznik inzulinové rezistence ve svalové buňce v důsledku nahromadění lipidových inkluzí jsou podobné s mechanismy, kterými negativně působí mastné kyseliny. Klíčovou roli zde sehrává interakce s nitrobuněčnými signalizačními kaskádami, které jsou aktivovány po navázání inzulinu na membránový receptor. Schopnost myocytů efektivně metabolizovat lipidy je dána také počtem mitochondrií a aktivitou mitochondriálních enzymů. U obézních pacientů a diabetiků 2. typu je oxidační kapacita svalové buňky snížená. Pro klinickou praxi je důležitý poznatek, že vhodný pohybový trénink vede u obézních jedinců a pacientů s diabetes mellitus 2. typu ke zvýšení počtu mitochondrií a větší oxidaci extra- i intracelulárně uložených lipidů, čímž se zlepšuje citlivost svalové buňky k účinkům inzulinu.
Tuková tkáň jako endokrinní orgán
V současnosti byla identifikována řada látek proteinové povahy, které jsou produkovány tukovou tkání. Tyto produkty jsou souhrnně označovány jako „adipokiny“ a na jejich produkci se vedle samotných adipocytů podílejí i makrofágy a další imunokompetentní buňky lokalizované v tukové tkáni. Některé z adipokinů mají efekt pouze parakrinní, většina adipokinů se ovšem dostává do krevního řečiště a působí pak typickým endokrinním způsobem ve vzdálených cílových orgánech (sval, játra, pankreas, mozek, endotelie).
Spektrem účinku představují velmi heterogenní skupinu podílející se na regulaci intermediárního metabolismu, centrální regulaci příjmu potravy, tkáňové utilizaci substrátů i regulaci inzulinové senzitivity, mají vztah k etiopatogenezi aterosklerózy a regulaci imunitních odpovědí. Obezita je charakterizována porušenými plazmatickými hladinami těchto cirkulujících adipokinů a rozvojem prozánětlivého stavu, který je asociován s rozvojem inzulinové rezistence. V následujícím textu uvedeme jako modelové zástupce tři adipokiny: adiponectin, leptin a tumor necrosis factor- α.
Adiponectin
Adiponectin je produkován téměř výlučně v adipocytech. V plazmě je přítomen v překvapivě velkém množství (koncentrace činí 2–20 mg/l) a tvoří mezi ostatními adipokiny výjimku v tom smyslu, že jeho plazmatická hladina je u osob s nadbytkem tukových zásob, diabetiků 2. typu a osob s ischemickou chorobou srdeční snížena, zatímco koncentrace ostatních adipokinů v plazmě jsou u těchto stavů většinou zvýšeny.
Adiponectin významným způsobem zasahuje do regulace sacharidového a lipidového metabolismu. Parenterální podání adiponectinu vede k výraznému zlepšení inzulinové rezistence na zvířecím modelu diabetu, dochází ke zvýšení utilizace a transportu glukózy a mastných kyselin ve svalových, jaterních i tukových buňkách. Zároveň adiponectin potlačuje glukoneogenezi v hepatocytech.
Metabolické a inzulin senzitizující účinky jsou na molekulární úrovni zprostředkovány aktivací nitrobuněčného enzymu AMP-kináza (adenosin-monofosfát aktivovaná kináza), který představuje jakousi centrální buněčnou energetickou výhybku detekující nedostatek či nadbytek energie pro buněčné procesy. Plazmatická koncentrace adiponectinu negativně koreluje s obsahem tuku v těle, BMI (body mass index), koncentrací triglyceridů, lačnou glykémií, lačnou hladinou inzulinu a různými parametry inzulinové rezistence. Naopak plazmatická hladina HDL cholesterolu je s hladinou adiponectinu asociována pozitivně.
Adiponectin zabraňuje transformaci makrofágů v pěnité buňky, snižuje expresi povrchových adhezních molekul na povrchu makrofágů a významně omezuje nežádoucí proliferaci buněk hladkého svalstva a vaziva během reparačních procesů. Účinně tak brání rozvoji a progresi aterosklerózy.
Leptin
Leptin je kromě tukové tkáně v menší míře produkován také v jiných tkáních (žaludek, játra, placenta, sval). Leptin má zásadní podíl v dlouhodobém udržování tělesné hmotnosti působením na hypothalamická centra regulující příjem potravy. Inhibuje zde neurony zodpovědné za příjem potravy a naopak stimuluje aktivitu sympatického autonomního nervového systému, čímž zvyšuje bazální energetický výdej.
U zdravých jedinců stoupá plazmatická koncentrace leptinu se zvyšujícím se množstvím tukové tkáně a skrze účinek na hypothalamická centra následně dochází zpětnovazebně k potlačení chuti k jídlu a zvýšení energetického výdeje. Leptin tak za fyziologických podmínek funguje jako dlouhodobý regulátor tukových zásob v organismu. U obézních jedinců ovšem dosud ne zcela jasnými mechanismy leptin ztrácí tuto schopnost, hovoří se o leptinové rezistenci.
Tumor necrosis factor-α
TNFαTNFα (tumor necrosis factor-α , kachexin) byl historicky prvním z řady adipokinů, který byl navržen jako spojující článek mezi obezitou a inzulinovou rezistencí. Je produkován zejména buňkami imunitního systému lokalizovanými v tukové tkáni. Na zvířecím modelu a in vitro indukuje TNFα inzulinovou rezistenci přímou interakcí s inzulinovou signalizační kaskádou. TNFα také mohutně aktivuje lipolýzu v tukové tkáni, čímž dochází ke zvýšení hladiny volných mastných kyselin v plazmě. U obézních pacientů je exprese genu pro TNFα v podkožní tukové tkáni zvýšena.
Shrnutí
Etiopatogeneze inzulinové rezistence a diabetes mellitus 2. typu není dosud plně objasněna. Poruchy v metabolismu tukové tkáně, zvýšená plazmatická hladina volných mastných kyselin a produkce hormonů v tukové tkáni představují možné mechanismy. Bude tedy úkolem pokračujícího výzkumu popsat další možné faktory podílející se na vzniku diabetes mellitus 2. typu.
|
|
Vybraná literatura 1. Trayhurn P and Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr 92: 347-355, 2004.
2. Boden G. Free fatty acids-the link between obesity and insulin resistance. Endocr Pract 7: 44-51, 2001.
3. Randle PJ. Regulatory interactions between lipids and carbohydrates: the glucose fatty acid cycle after 35 years. Diabetes Metab Rev 14: 263-283, 1998
4. Hotamisligil GS. Inflammation and metabolic disorders. Nature 444: 860-867, 2006.
5. Lafontan M and Berlan M. Fat cell adrenergic receptors and the control of white and brown fat cell function. J Lipid Res 34: 1057-1091, 1993.
6. Trujillo ME and Scherer PE. Adipose tissue-derived factors: impact on health and disease. Endocr Rev 27: 762-778, 2006.
7. Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA, Bogardus C, Jenkins AB and Storlien LH. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 46: 983- 988, 1997.
8. Hansen L, Pedersen O. Genetics of type 2 diabetes mellitus: status and perspectives. Diabetes Obes Metab. 2005 Mar;7(2):122-35.
9. Muoio DM , Newgard CB: Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat.Rev Mol.Cell Biol. 9(3), 193-205 (2008).
10. Guilherme A, Virbasius JV, Puri V, Czech MP: Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat.Rev Mol.Cell Biol. 9(5), 367-377 (2008).
11. Jeukendrup AE: Regulation of fat metabolism in skeletal muscle. Ann.N.Y.Acad.Sci. 967, 217-235 (2002).
12. Berg AH and Scherer PE. Adipose tissue, inflammation, and cardiovascular disease.Circ Res 96: 939-949, 2005.
13. Bergman RN and Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus Trends Endocrinol Metab 11: 351-356, 2000.
14. Stich V, Berlan M. Physiological regulation of NEFA availability: lipolysis pathway. Proc Nutr Soc. 2004 May;63(2):369-74. Review
(Plnou verzi článku najdete ne www.sanquis.cz v rubrice SANQUIS PLUS - medicína)
Celý článek ve formátu pdf naleznete zde.
|
|
|
|
obsah čísla 59 |
 |
ročník 2008 |
|
témata |
|
|